iCLIP.meta.processing_index¶
-
iCLIP.meta.processing_index(interval_iterator, bam, window_size=50)¶ Calculate the ratio of processed transcripts to non-processed
- interval_iterator : CGAT.Bed or CGAT.GTF-like iterator
- The iterator must yeild objects that have a start, end and strand attribute. Processing index will be calculated around these.
- bam : *_getter-like function
- A getter function returned by the make_getter function, this will be used to retrieve cross-link counts.
- window_size : int, optional
- How far up and downstream of the the processing site to consider.
- int
- processing index averaged over all processing sites given.
The proccessing index for G genes is defined as:
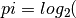
rac{sum_{i=1}^{G} N_i^{PM}}{sum_{i=1}^{G} N_i^M})
after Baejen et al Mol Cell 5(55):745-757. However, Beaejen et al normalise this number to the total number of genes, which seemswrong to me.