iCLIP.kmers.pentamer_enrichment¶
-
iCLIP.kmers.pentamer_enrichment(gtf_chunk_iterator, bam, fasta, kmer_length=5, randomisations=100, seperate_UTRs=False, spread=15, pool=None)¶ - This function calculates the z-score of enrichment of kmers around
cross-linked sites.
- gtf_chunk_iterator : iter of list-like of CGAT.GTF.Entry
- An iterator that returns a list-like object containing CGAT.GTF.Entry objects representing a gene or transcript.
- bam : *_getter func
- A getter function, as returned by
make_getter() - fasta : CGAT.IndexedFasta
- Indexed fasta containing the genome sequence to get the transcript sequences from.
- kmer_length : int, optional
- length of kmer to search for enrichment of. All possible kmers of this length will be tested. Defaults to 5.
- randomisations : int, optional
- Number of sequence randomisation to perform in order to determined mean and sd of the frequency of each kmer under a the null model.
- seperate_UTRs : bool, optional
- No effect to be removed
- spread : int, optional
- Number of bases to consider around a clip site when calculating an overlap. Defaults to 15.
- pool : multiprocessing.Pool, optional
- If present, work will be parallelized across the worker pool
- pandas.Series of float
- z-values of the frequency of overlaps between cross-link sites and all kmers. Series is the kmer in question
For each transcript/gene/collection of intervals in gtf_chunk_iterator, the count of cross-link sites is calculated. Each cross-link site is then extended by spread bases in both directions.
For every possible kmer, the start positions in the sequence of the transcript is then calculated and the number of times a cross-link site overlaps a kmer start position is counted.
The positions of the cross-link site are then randomized. And the above process is repeated on each of the randomized profiles.
This is performed on introns and exons seperately.
Once this has been performed on all transcripts/genes in the iterator, scores are summed across all transcripts/genes and the mean and standard deviation of the counts for each kmer is calculated across the randomisations. For each kmer
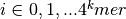 the
z-score
the
z-score 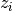 is calculated:
is calculated: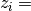
rac{observed_counts - mu_i}{sigma_i}
where
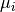 is the mean frequency of overlaps between
cross-links and the i*th* kmer across the randomisations and
is the mean frequency of overlaps between
cross-links and the i*th* kmer across the randomisations and
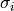 is the corresponding standard deviation.
is the corresponding standard deviation.This process can be very time consuming for longer kmers. To help elleviate this, it can be parrellized accross cores of the machine by providing a multiprocessing.Pool object to the pool parameter. The task will then be parrellized, gene-wise.